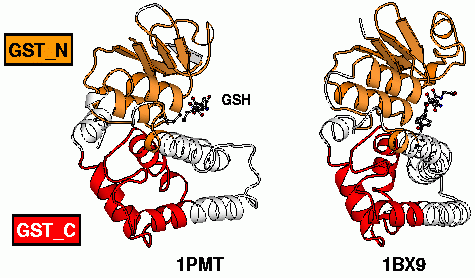
| Example of two glutathione S-transferases. The two most conserved parts among various GSTs were highlighted. |
| Example of two glutathione S-transferases. The two most conserved parts among various GSTs were highlighted. |
Glutathione S-transferases (GSTs) constitute a large family of proteins. Historically, GSTs were divided into several classes based on biochemical and sequence considerations. It is admitted that all GSTs have a conserved 3D-structure, and indeed, more than twenty different GSTs from various classes were crystallized, confirming this assertion.
The table below presents a selection of GSTs for which X-ray structures are available. A nickname was given to most of these sequences to simplify their manipulation.
| nickname | PDB-ID | SwissProt-ID | Description |
|---|---|---|---|
| alpha1 | 1guh | GTA1_HUMAN | Class alpha, with S-benzyl-glutathione as ligand. |
| alpha2 | 1gul | GTA4_HUMAN | Class alpha with iodobenzyl glutathione as ligand. |
| alpha3 | 1guk | GTA4_MOUSE | Class alpha with iodobenzyl glutathione as ligand. |
| alpha4 | 1fhe | GT27_FASHE | Class alpha, ligand: glutathione |
| alpha5 | 1gta | GT26_SCHJA | Class alpha, ligand-free. ligand: none |
| beta1 | 1a0f | GT_ECOLI | Class beta, with glutathionesulfonic acid as ligand. |
| beta2 | 2pmt | GT_PROMI | Class beta, with glutathion as ligand. |
| phi1 | 1axd | GTH1_MAIZE | Class phi, ligand: actoylglutathione |
| phi2 | 1gnw | GTH4_ARATH | Class phi with S-hexylglutathione as ligand. |
| mu1 | 1gtu | GTM1_HUMAN | Class mu, ligand-free. |
| mu2 | 2gtu | GTM2_HUMAN | Class mu, ligand free. |
| mu3 | 2gst | GTM1_RAT | Class mu ligand: GPS + Sulphate |
| mu4 | 1gsu | GTM2_CHICK | Class mu with S-hexylglutathione as ligand. |
| omega | 1eem | tn:AAF73376 | Omega, ligand: glutathione + 2 sulfate ions |
| pi1 | 1glp | GTP1_MOUSE | Class pi, ligand: glutathione sulfonic acid |
| pi2 | 2gsr | GTP_PIG | Class pi with ligand: ILG-OCS-GLY |
| pi3 | 2gss | GTP_HUMAN | Class pi with ethacrynic acid as ligand. |
| sigma | 2gsq | GTS_OMMSL | Class sigma with s-(3-iodobenzyl)glutathione as ligand. |
| theta | 1ljr | GTT2_HUMAN | Class theta, with glutathione as ligand. |
| zeta | 1e6b | Q9ZVQ3 | Class Zeta. |
| ure2 | 1hqo | URE2_YEAST | Nitrogen regulation fragment of the yeast prion protein ure2p. |
| clic | 1k0m | CLI1_HUMAN | Soluble form of the intracellular chloride ion channel Clic1. |
The t_coffee documentation is available online from the t_coffee home page. Please refer to this documentation for explanations about the many switches employed in the exercises below.
We will restrict our attention to a selection of GSTs that encompass the diversity of the sequences for which a structure is available. Note that many GSTs, especially the bacterial ones, belong to other new classes yet to be described.
Let us define our test set with the help of an environmentstart by downloading all the needed files: FILES
gunzip gst_exercise_files.tar.gz
tar -xvf gst_exercise_files.tar
cd gst_exercise_files
bash
export TEST="alpha1.pdb beta1.pdb phi1.pdb mu1.pdb omega.pdb pi1.pdb \
sigma.pdb theta.pdb ure2.pdb clic.pdb"
This last command defines the sequences on which you will make the analysis
Note that these "pdb" files are uncomplete: they only contain the alpha atoms of a selected single chain. They were produced with the Perl script extract_from_pdb which is distributed with t_coffee.
First, we want to produce three libraries for t_coffee. We'll use these libraries to build multiple sequence alignment in the next exercises. This strategy is intended to demonstrate the versatility of t_coffee and also save some CPU time. Note that t_coffee extracts the amino acid sequences directly from the pdb file, but one could also have supplied these sequences in FASTA format, where the structural information was irrelevant. Let us build the libraries:
The method fast_pair produces a global alignment for every possible pair of sequences. The FASTA heuristic is used (other global methods are available).
t_coffee \ -in $TEST Mfast_pair \ -out_lib gst_fast_pair.lib \ -quiet stdout \ -convert
The library was saved in the file gst_fast_pair.lib. Have a look at the content of this file.
The method lalign_id_pair computes the ten best local alignments for every pair of sequences using the Smith-Waterman algorithm. This is more CPU expensive than fast_pair.
t_coffee \ -in $TEST Mlalign_id_pair \ -out_lib gst_lalign_id_pair.lib \ -quiet stdout \ -convert
Have a look at the gst_lalign_id_pair.lib file. Can you see how its content differs from that of the gst_fast_pair.lib file.
The method sap_pair calls the external program SAP which performs a structure-based alignment for every pair of structures. Only the coordinates of the alpha atoms are taken into account (the type of residue is ignored). The structures are allowed to have some flexibility during the alignment process. Because this method is pretty CPU expensive, the library gst_sap_pair.lib has been precomputed.
t_coffee \ -in $TEST Msap_pair \ -out_lib gst_sap_pair.lib \ -quiet stdout \ -convert
We will now employ the libraries to produce a multiple sequence alignment (MSA). As a proof of principle, let's start by making a very naive MSA by exploiting the information of the global library only:
t_coffee \
-in Lgst_fast_pair.lib \
-run_name global \
-outorder input \
-clean_aln 0 \
-output clustalw_aln score_html
This creates two files: global.clustalw_aln that contains the alignment in text format, and global.score_html in html format (postscript and pdf output are also available). Have a look at the score_html file: The color scale denotes the consistency of a residue, i.e. how well its position in the MSA is supported by the supplied libraries. The color scale is not related to the degree of conservation of the column in the alignment. A warm color (red to orange) indicates that the position of a residue in the MSA is well supported, a cold color, (green to blue) indicates that the position of a residue in the the MSA is poorly or not supported by the library.
There are clearly two "redish" blocks that are visible in this alignment, and which contain the few fully conserved residue. In the greenish parts of the alignment appear a few oddities, some regions in the middle and at the C-terminus, which do not form "clean" blocks. It is possible to improve the appearance of this MSA by allowing t_coffee to rearrange the residues with low consensus score, for example using
t_coffee \
-in Lgst_fast_pair.lib \
-run_name clean_global \
-outorder input \
-clean_aln 1 \
-clean_threshold 2 \
-clean_iteration 5 \
-output clustalw_aln score_html
Compare clean_global.score_html with global.score_html: Although the cleaned alignment looks better, the displaced residues are not supported anymore by the used library. This is only a cosmetic change and there is no biological or scientific argument to support it. Beware of nice-looking alignments!
This is how to mix the two libraries of global and local sequence alignments
t_coffee \
-in Lgst_fast_pair.lib Lgst_lalign_id_pair.lib \
-run_name default \
-outorder input \
-clean_aln 0 \
-output clustalw_aln score_html
When compared with the previous global-only example, the overall consistency slightly decreases, which is linked to the fact that most alignements in the local library are random alignents. One can also observe a few differences in the alignments as compared to the previous one, which could be difficult to justify/evaluate at this stage.
This strategy of mixing the local and the global sequence library is in fact the default for t_coffee on a set of sequences. Indeed, the whole process of making the local and global libraries and then running the above command can be simply realized by the command
t_coffee global.clustalw_aln -outorder input -clean_aln 0
Note that global.clustalw_aln is used here to supply the sequences (the gaps are ignored). Another simple command yields the same result
t_coffee -in $TEST Mlalign_id_pair Mfast_pair -outorder input -clean_aln 0
So now let us build the alignment from the structural library alone
t_coffee \
-in Lgst_sap_pair.lib \
-run_name sap \
-outorder input \
-clean_aln 0 \
-output clustalw_aln score_html
One can easily recognize in the sap.score_htlml file, the core regions of the GSTs that were repeatedly (correctly) aligned by SAP (warm colors), and the more flexible loop regions where no consensus alignement emerged (in blue).
Note that that two short but highly consistent stretches at the N-terminus actually correspond to the active sites of GSTs.
Who could now resist from building an alignment with the three libraries?
t_coffee \
-in Lgst_lalign_id_pair.lib Lgst_fast_pair.lib Lgst_sap_pair.lib \
-run_name all \
-outorder input \
-clean_aln 0 \
-output clustalw_aln score_html
The latter alignment ressembles to the structure-only alignment. Why?
Apart from creating libraries and assembling them into MSA, t_coffee also permits us to evaluate a MSA in the light of another library. Let us use this feature to decipher the respective contribution of the sequence and structural information in the last example:
t_coffee all.clustalw_aln \
-in Lgst_lalign_id_pair.lib Lgst_fast_pair.lib \
-score \
-quiet stdout \
-outorder input \
-clean_aln 0 \
-run_name all_vs_seq \
-output score_html
t_coffee all.clustalw_aln \
-in Lgst_sap_pair.lib \
-score \
-quiet stdout \
-outorder input \
-clean_aln 0 \
-run_name all_vs_struct \
-output score_html
Compare all_vs_seq.score_html with all_vs_struct.score_html.
In the light of the structure-based MSA, there is one obvious mistake at the N-terminus of the default alignement: the tyrosine 26 of the omega GST is not correctly aligned, for example with the histidine 6 of the sigma GST. Using your favorite text editor produce a very small library (name it gst_active_site.lib) with just a few pairs in order to correct this aspect of the default alignement running the command
t_coffee \
-in Lgst_fast_pair.lib Lgst_lalign_id_pair.lib Lgst_active_site.lib\
-run_name test \
-outorder input \
-clean_aln 0 \
-output clustalw_aln score_html
Choose three structures and produce a library using the method sap_pair. Then, re-align all GSTs using the complete local and local sequence libraries, and the partial structural library. Do three structures suffice to improve the alignment?